Foundation models for cell and tissue imaging are clearing the benchmarks. Whether they are clearing the right benchmarks is a different question.
The term “foundation model” has entered bioimaging, and with it a promise of generality: a single pretrained model that segments any cell, or a whole-slide encoder that produces features suitable for any downstream task, with far less local annotation than before. Two developments over the last year made that promise concrete. In cell segmentation, Cellpose-SAM reports that it “substantially outperforms inter-human agreement and approaches the human-consensus bound” and, on that basis, positions itself as a foundation model for biological segmentation [4]. In pathology, a wave of whole-slide models (UNI, Prov-GigaPath, Virchow, H-optimus) now sits above the ResNet/ImageNet generation on essentially every public leaderboard [12, 13].
Both claims are true. Neither settles the question that matters before building a pipeline on top of these models.
The problem is not hype in the ordinary sense. These models are real progress. The problem is subtler: the metric that certifies a model as “superhuman” or “state of the art” is frequently not the metric that governs whether the biological conclusion drawn from its output is correct. The certifying benchmark and the scientific readout have diverged. Once that gap is visible, the relevant question is no longer “is this a foundation model?” but “is it superhuman at the specific quantity a given result depends on?”. That question is local and empirical, and no leaderboard answers it.
The genuine advances
Generalist segmentation has changed the first pass of a bioimage workflow. Cellpose, StarDist and Mesmer already turned segmentation into a starting point rather than a from-scratch build [1, 2]; the SAM-backbone generation extended out-of-the-box generalization further, and, most useful in practice, Cellpose-SAM was explicitly hardened against channel shuffling, cell-size variation, shot noise, downsampling and blur [4]. These are ordinary conditions in a multiplexed imaging core, not academic perturbations: inconsistent panels, uneven staining, out-of-focus tiles. Coupling restoration directly to segmentation, as Cellpose3 does, is a sound design choice because it optimizes the image for the measurement that follows rather than for visual inspection [3].
Whole-slide pathology models show a similar pattern. Pretraining on hundreds of thousands to millions of slides produces embeddings that outperform the previous generation across tissue classification, biomarker prediction and subtyping [12, 13]. The labor savings are substantial: fewer local annotations and a stronger starting representation.
The critique that follows is therefore not that these models do not work. It is that the reported metric does not measure the quantity that matters.
At the cell level: the mask is not the measurement
Segmentation quality is almost universally reported as mask overlap, an IoU-based F1 score conventionally thresholded at 0.5. A model that scores well by this measure is called accurate. But the mask is not the deliverable. The deliverable is everything computed from the mask: the per-cell expression profile, the phenotype call, the neighborhood, the niche.
Bruhns and colleagues quantified this in a recent benchmark on multiplexed tissue imaging [6]. Using controlled, realistic perturbations of ground-truth segmentations, they show that even moderate errors distort estimated protein profiles and disrupt cellular neighborhood relationships in feature space, that Gaussian-mixture phenotyping begins to misclassify closely related cell types, and that the effect extends to the inferred cell lineage, not only fine subtypes. The central result is this: clustering agreement with ground truth does not reach perfect agreement even when the segmentation’s IoU-F1 score reaches 100. The reason is structural. A 0.5 IoU threshold allows a substantially mis-segmented cell to count as “correct,” so cells that pass the metric still carry enough boundary error to shift their features, and therefore their cluster assignment. A segmentation can be “perfect” by the certifying metric and still alter the resulting biology.
This is not an isolated finding. In spatial transcriptomics, Mitchel and colleagues report that segmentation-driven artifacts frequently dominate downstream results such as differential expression, neighborhood influence and ligand-receptor inference, to the point where the top “hits” are contaminating signal from adjacent cells rather than biology [7]. The mechanism differs (molecule mis-assignment rather than boundary overlap), but the conclusion is the same. A step often treated as routine preprocessing can be a leading source of false signal, and the metric used to validate it does not register the failure.
The practical consequence is concrete. A “superhuman” segmentation model, validated on masks, can still produce a feature table that is wrong precisely where boundaries are hardest to draw, often for the small immune cells, which tend to suffer the largest proportional boundary error.
At the slide level: the site-signature problem
At the level of whole-slide pathology models, the same gap reappears in a starker form. Here the certifying metric is downstream task performance, typically AUC on a classification or biomarker benchmark. What that score does not reveal is which features the model relies on to achieve it.
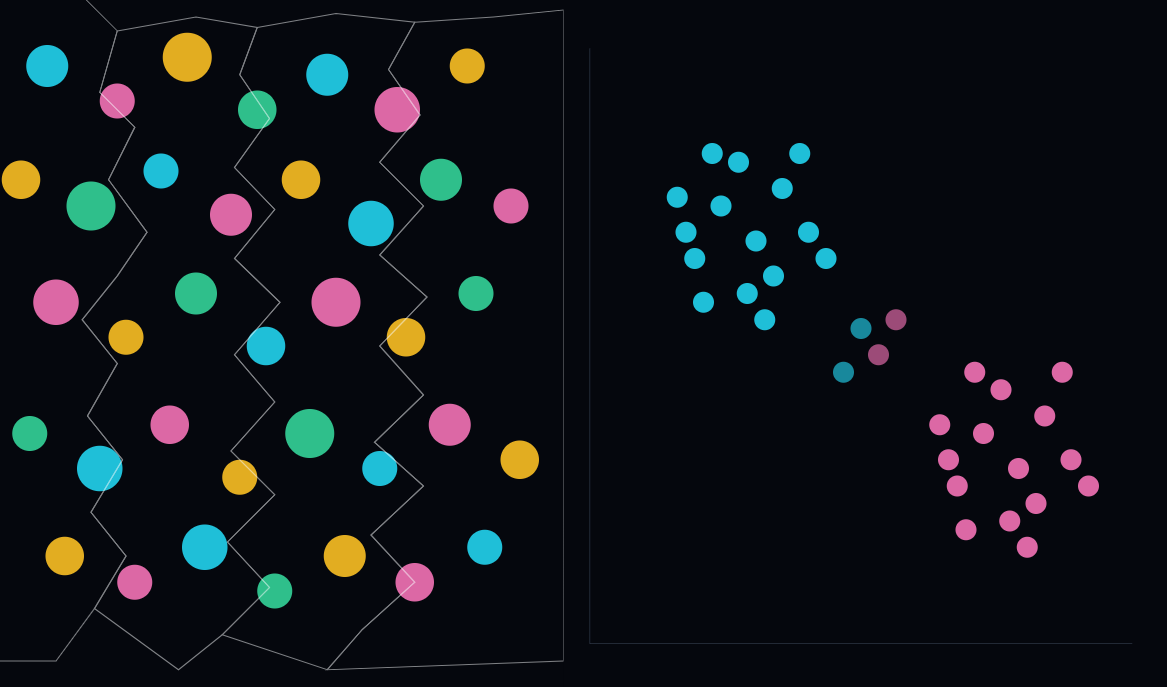
de Jong and colleagues examined this directly [8]. They introduce a Robustness Index measuring whether a model’s embedding is organized by biology or by the confounding “signature” of the medical center: the staining protocol, the scanner, the fixation. Across ten public pathology foundation models, all encode the medical center to a strong degree. Only one has a robustness index above one, meaning its biological signal only marginally exceeds the site signature. In the embedding spaces they examine, tissue clusters by hospital more than by cancer type, and the center of origin is predicted more accurately than the biology. The downstream errors are not random: cancer-type misclassifications are specifically attributable to same-center confounders. A model can top an in-distribution benchmark while partly recognizing where the slide was produced rather than the underlying pathology. This is a current instance of an older, well-documented problem, namely that networks can predict the TCGA acquisition site from the image alone [10].
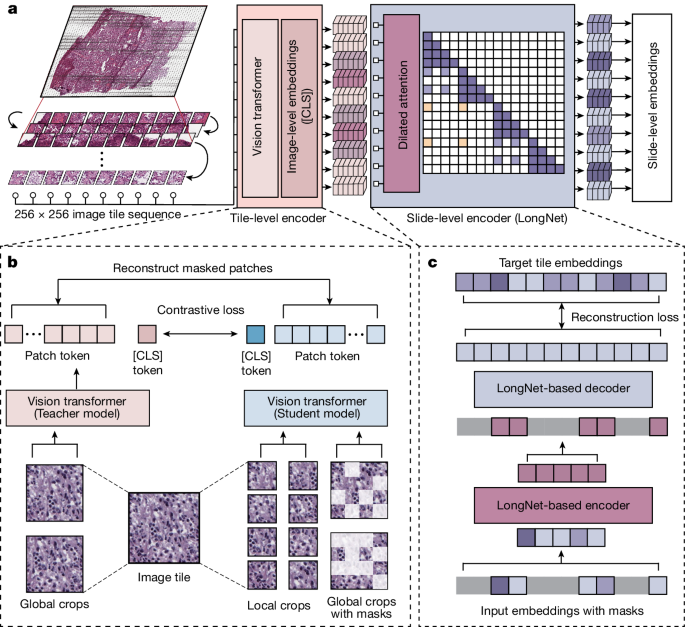
The Prov-GigaPath whole-slide pathology foundation model. (a) A whole-slide image is divided into 256×256 tiles; a tile encoder embeds each tile, and a LongNet-based slide encoder aggregates those embeddings into slide-level features for downstream tasks. (b) The tile encoder is pretrained with DINOv2 self-supervised learning. (c) The slide encoder is pretrained with a masked-autoencoder objective built on LongNet. Adapted from Xu et al., Nature 630, 181–188 (2024), CC BY 4.0.
Scale does not resolve this, contrary to what the foundation-model narrative implies. Comprehensive benchmarking finds that model size and training-set size do not consistently track downstream performance in pathology [11]; models trained on an order of magnitude fewer slides can match far larger models on standard tasks. Whatever the benchmark rewards, it is not a simple function of more data and larger models, which is precisely the assumption the term “foundation” encourages.
The relocation of expertise
Taken together, the two levels point to a single principle. In both cases the model is validated against an in-distribution, proxy target (mask overlap, benchmark AUC), while the quantity that matters lies elsewhere: the correctness of the phenotype and neighborhood, and the model’s behavior on a new center, scanner, panel or tissue it has not seen. The certifying metric and the scientific readout are different objects, and the foundation-model label obscures the difference.
For anyone deploying these tools, that reframes the work. It does not mean avoiding foundation models. They are the right starting point. It means moving the unit of validation downstream and out-of-distribution:
- Downstream evaluation. A segmentation is more usefully judged by the stability of the feature table and phenotype assignments it produces than by mask IoU alone. A slide encoder is more usefully judged on data from a held-out center than on a random in-distribution split.
- Site confounding. A center or scanner signature is best assumed present unless a test shows otherwise. A center- or scanner-stratified holdout permits a direct check of whether a downstream classifier is separating biology from batch; where the center of origin is recoverable from the embedding, the downstream model can exploit it.
- Correction and annotation as pipeline cost. Segmentation-error correction, quality control, and a limited amount of local fine-tuning are ordinary components of the analysis cost rather than remedial exceptions. In most cases they are what separates a trustworthy measurement from a strong prior.
- Maturity of the evidence. Several of the results cited here, including Cellpose-SAM, remain preprints [4, 8, 9, 11, 14] and have not completed peer review.
The foundation-model era has not removed the need for image-analysis expertise; it has relocated it, from building the model to auditing it. The work is no longer training a bespoke network for one panel. It is deciding which of a model’s superhuman numbers to trust for a given tissue and question, a judgment that cannot be pretrained.
The relevant question is not whether a model is a foundation model, but whether it is superhuman at the specific quantity a given conclusion depends on. Answering that is a local, empirical matter, and one that still calls for expert judgment.
Milad Adibi
References
- Stringer C, Wang T, Michaelos M, Pachitariu M. Cellpose: a generalist algorithm for cellular segmentation. Nature Methods (2021) 18:100-106.
- Greenwald NF, et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning (Mesmer/TissueNet). Nature Biotechnology (2022).
- Stringer C, Pachitariu M. Cellpose3: one-click image restoration for improved cellular segmentation. Nature Methods (2025).
- Pachitariu M, Rariden M, Stringer C. Cellpose-SAM: superhuman generalization for cellular segmentation. bioRxiv 2025.04.28.651001 (2025). [preprint]
- Archit A, et al. Segment Anything for Microscopy (µSAM). Nature Methods (2025).
- Bruhns M, Schleicher JT, Wirth M, Zago M, Babaei S, Claassen M. Effects of segmentation errors on downstream-analysis in highly-multiplexed tissue imaging. PLoS Computational Biology (2025) 21(9):e1013350.
- Mitchel J, et al. Impact and correction of segmentation errors in spatial transcriptomics. Nature Genetics (2025). doi:10.1038/s41588-025-02497-4.
- de Jong ED, Marcus E, Teuwen J. Current pathology foundation models are unrobust to medical center differences. arXiv:2501.18055 (2025). [preprint]
- Kömen J, Marienwald H, Dippel J, Hense J. Do histopathological foundation models eliminate batch effects? A comparative study. arXiv:2411.05489 (2024). [preprint]
- Dehkharghanian T, et al. Biased data, biased AI: deep networks predict the acquisition site of TCGA images. Diagnostic Pathology (2023) 18:67.
- Bareja R, et al. Evaluating vision and pathology foundation models for computational pathology: a comprehensive benchmark study. medRxiv 2025.05.08.25327250 (2025). [preprint]
- Chen RJ, et al. Towards a general-purpose foundation model for computational pathology (UNI). Nature Medicine (2024).
- Xu H, et al. A whole-slide foundation model for digital pathology from real-world data (Prov-GigaPath). Nature (2024) 630:181-188.
- Neidlinger P, et al. Benchmarking foundation models as feature extractors for weakly-supervised computational pathology. arXiv:2408.15823 (2024). [preprint]