
When you want to learn more about astronomy, do you read a book about the automobile industry? No, you select the most accurate and up-to-date map of the celestial objects. To explore a species genome, the most accurate reference needs to be used to perform the best analysis. Despite the recent improvement in genome sequencing, all the pharmaceutical researches are still using the human reference genome from 2013. Last year, a new and complete version of a human genome was published.
In the previous version of the human genome, at least 8% remained unknown. On the contrary, in this new version, 100% of the genome is explored ! Which one would you use?

Until recently, even still, we are using a human genome from 2013, patched in 2019 (GRCh38.p13), generated by the Genome Reference Consortium (GRC). Unfortunately this version of the human genome is not complete and presents 230 mega-base pairs (Mbp) of issues. This lack of information is mainly due to the technique used to generate the human reference genome. We cloned human DNA into bacteria to construct this genome. However, such technique does not allow the exploration of repetitive sequences which are estimated to 151 Mbp, which are spread throughout the human genome.
But recently the human genome was improved (Nurk et al., 2022). This new version, called T2T-CHM13, completed the remaining 8 % of the unknown sequence in 20 years. It benefited from new technologies: the ultra long-read sequencing, from PacBio and Oxford Nanopore which respectively resolved the gaps (not contiguous sequences) that were present in the GRCh38.p13 and permitted the complete assemblies of centromere and even chromosome. But these two long-read sequencing technologies present a high error rate. However another technology from PacBio, the most recent “HiFi” sequences long reads (15-20 kbp) at a compromise error rate. This technology permits them to explore the repetitiveness of the human genome, which represents more than 50% of it.
After performing the sequencing, scientists proceeded to genome assembly. Most of the chromosomes resulted in a single sequence, assessing the accuracy of the sequencing technologies used. However, chromosome 9 presents a repetitive region preventing a linear assembly (top right); and chromosomes 13, 14, 15, 21, and 22 are connected and present highly similar content (bottom right). This similarity is due to the ribosomal RNAs that encode the 45S rRNA and are arranged in large tandem on the genome, making them harder to explore. With more than 300 copies of these sequences in a human genome, their assembly needed the construction of more complex algorithms using sparse de Bruijn graphs to reduce the overlaps in genome assembly.
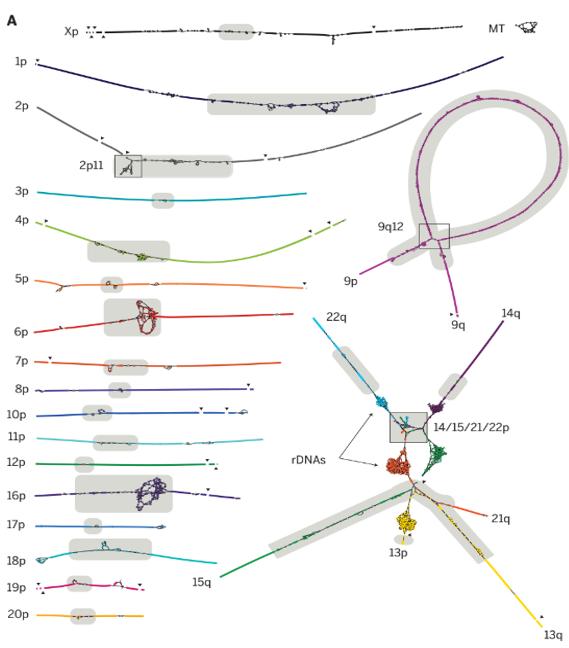
In order to validate the genome assemblies, all the individual sequenced reads were aligned to the generated CHM13 draft assembly, by exploring the variants. This step resulted in the assembly: T2T-CHM13v1.1. Sequence genome is uniformly covered, as expected for high quality genome assemblies. The accuracy is estimated to be one error per 10 Mbp ! Compared to the previous version, which presented 8% of unknown sequences, the T2T-CHM13 genome is covered by only 0,3% errors !
3,054,832,041 bp combine the 22 autosomes, the chromosome X, and the mitochondrial chromosome.
The next step for genome assembly is to annotate the sequence by exploring the genes, known and unknown present on the sequence.
Scientists compared the already known human genome annotation from the previous version to the T2T-CHM13. To explore further they sequenced the transcriptome using a full-length technology, they de novo assembled the transcripts then compared to the genome assembly. This exploration permits to generate 5% more genes than previous annotation. Most of the genes are non-coding and only 19,969 genes are predicted to be protein coding genes. 3,604 genes seem specific to CHM13.
If you want to know more about this magnificent project, you can read this Science article or one of the even more detailed articles presented in this special issue of the Science journal.
Genome individuality represents only 1% of each individual genome. Ideally, in order to perform even more accurate genome analysis, one would need to sequence with high quality thousands of genomes to explore in detail the great diversity of our genomes !
References
Jane Alfred and Ian T Baldwin, The Natural History of Model Organisms: New opportunities
at the wild frontier. eLife 4:e06956 (2015). DOI:10.7554/eLife.0695
Sergey Nurk et al., The complete sequence of a human genome. Science 376, 44-53 (2022).
DOI:10.1126/science.abj6987
Heather Turner, Clinical long-read sequencing (2022). PHG Foundation.
https://www.phgfoundation.org/briefing/clinical-long-read-sequencing